
 Yahoo Finance
Yahoo Finance New Muscle Disease Indication for ATL1102 – Limb Girdle Muscular Dystrophy R2

Figure 1
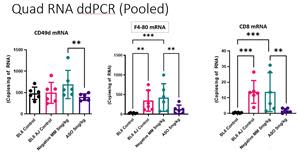
Figure 2
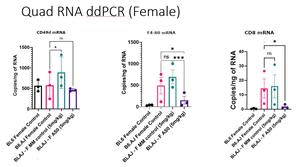
LGMDR2, also referred to as dysferlinopathy, is a rare genetic muscle disease caused by mutations in the dysferlin gene (DYSF)
Animal study conducted at MCRI in collaboration with Jain Foundation in the US
Study results showed significant decreases in target CD49d RNA and key immune cell RNA levels in the muscle
Data supports progression into a longer-term efficacy study planned for 3Q/4Q2022
Mark Diamond, Antisense Therapeutics Managing Director and CEO said: “It is exciting for the Company to be announcing a successful scientific exploration into a new muscle disease indication for ATL1102, LGMDR2. This was a strategically minded move on behalf of the Company to capitalise on the extensive data package generated to date on ATL1102 and to broaden ANP’s product pipeline. Similar to DMD, this is a rare muscle disease with a clear need for an effective treatment as emphasised by our expert consultants in the field. The results achieved in this first LGMDR2 animal study are showing the requisite effects on targeted immune cells. Having demonstrated target activity in a dysferlin animal model gives us confidence to advance the program in LGMDR2 and consider other muscle diseases with immune cell involvement as the data continues to point to the broader potential of ATL1102 as a treatment for such conditions.”
New York, NY, June 22, 2022 (GLOBE NEWSWIRE) -- Antisense Therapeutics Limited [ASX:ANP | US OTC:ATHJY | FSE:AWY] today announced the results from a first study of antisense to CD49d in a limb girdle muscular dystrophy R2 (LGMDR2) mouse model. LGMDR2 is a rare genetic muscle disease that is caused by mutations in the dysferlin gene that leads to significant reduction or absence of dysferlin protein levels in muscle fibers. Dysferlin loss occurs in both males and females with the condition called dysferlinopathy or LGMDR2. LGMDR2 is characterized by muscle inflammation, fibrosis, adiposity (fat) and progressive weakness in the hip and shoulder area (i.e. the limb girdle) proximal muscles (those closest to the center of the body) with loss of ambulation and upper limb function in adulthood. LGMDR2 affects ~ 1 in 125,000 people.1,2,3 There are no disease modifying agents in advanced development and no treatments have proven to be beneficial to slow the progression of the disease.
This first study of antisense to CD49d in the LGMDR2 mouse model (Bla/J mice with dysferlin loss) was undertaken in collaboration with experts in genetic muscle disease at the Murdoch Children’s Research Institute (MCRI) in Melbourne and the Jain Foundation in the USA. The Jain Foundation (https://www.jain-foundation.org/), a non-profit disease foundation established in the hopes of curing dysferlinopathy, is coordinating the worldwide efforts to find a treatment for dysferlinopathy and have substantial experience with LGMDR2.
In the study, Bla/J mice were treated with an antisense oligonucleotide (ASO) to CD49d at three dose levels (5, 10 and 20mg/kg) once weekly for six weeks, with a corresponding negative control oligonucleotide at the same dose levels. The study also included untreated Bla/J mice and healthy mice as controls. The results showed that at the low 5mg/kg/week ASO dose, the RNA levels of CD49d, and CD8 T cells and F4/80 macrophage cells in the quadricep muscle were significantly decreased by 42%, 86% and 70% respectively, compared to the levels observed in the mice dosed at the 5mg/kg/week dose level of the control oligonucleotide (P<0.05 Fisher’s Least Significant difference test). A significant reduction in CD8 RNA vs the untreated control Bla/J was also observed (see Figure 1 below).
Notably, in the quadricep muscle of the female untreated control mice, there was 2.5 times more F4/80 RNA than male mice (data on file). F4/80 is a marker of macrophages which are key immune cells involved in the disease and the increase in these cells is suggestive of a more active disease in the female mice. ASO treatment in female mice achieved significant reduction in F4/80 and CD8 cell mRNA in the quadricep muscle. In these same female mice, a trend towards reduction of 38% (P=0.0656) in CD49d RNA was observed compared to untreated Bla/J in another proximal muscle called the Psoas (see Figure 2 below). Given these observations the next study will be conducted in the female mice as agreed with the experts at the Jain Foundation.
The results from this first investigation of the potential of an antisense to CD49d drug in the Bla/J dysferlin deficient mouse model have shown the use of a low dose of the drug reduces the target (CD49d) and key immune cell (F4/80 macrophage and CD8+ T cells) RNA in the muscle. The results support the Company’s plans to move forward with the second phase (chronic setting) of the program with a follow on study in the same mouse model to test the potential of the low dose to reduce adipose (fat) levels, muscle loss and damage. The second study is planned for 3Q/4QCY22 (pending the availability of suitably aged mice) and designed to run for four months, with results to follow shortly thereafter.
The use of ATL1102 as a treatment for dysferlinopathy is covered in ANP’s patent application PCTAU2020/050445 directed at modifying muscle performance by reducing muscle adiposity. The recently filed provisional application 2021903024 also claims the use of ATL1102 to reduce thrombospondin-1 reported to be beneficial in treating the disease. The data from Bla/J mice studies can be used to support the prosecution of these claims and the filing of a new patent application.
Dr George Tachas Ph.D., Director, Drug Discovery and Patents at Antisense Therapeutics said “We are pleased to be continuing our key scientific collaboration with the MCRI while also benefiting from the expert advice provided by the Jain Foundation. The positive results from this first study in dysferlin deficient mice encourage us to move into the chronic phase of the program and we look forward with great anticipation to the results of this follow on study. There are several genetic muscle diseases with similar immune cell involvement like in LGMDR2 and DMD and so this data points to the potential use of low dose of ATL1102 as a treatment for such conditions.”
Laura Rufibach, Co-President of the Jain Foundation, said “With a singularly focused mission to find a cure for dysferlinopathy, we at the Jain Foundation are pleased to be working with Antisense Therapeutics and the MCRI on their research program evaluating the antisense to CD49d drug as a possible treatment of dysferlinopathy. We are encouraged by the results of the first study and look forward to the data of the longer dosing study to assess the efficacy of the treatment in mice in order to determine the potential of ATL1102 to be an effective therapy in this devastating and debilitating disease.”
Associate Professor Alastair Corbett, Neurologist at the Concord Repatriation General Hospital Neurology Clinic, said “LGMDR2 is a rare recessive muscular dystrophy with no effective treatment. The disease starts in late adolescence and patients show signs of muscle weakness at 15-30 years of age and it progresses slowly as people live into adult life. Patients exhibit weakness of the shoulder and hip muscles which adversely effects their upper and lower limb function. Sufferers of the disease struggle with mobility with a significant loss of quality of life and ultimately lose ambulation as they become wheelchair bound. Many muscle dystrophies with significant inflammation are in need of more effective therapies. I am very pleased to be involved with the hope that such endeavors may help a population in great need of an effective treatment.”
This announcement has been authorised for release by the Board.
For more information please contact:
Antisense Therapeutics | Investment Enquiries | US/European IR & Media |
Mark Diamond | Gennadi Koutchin | Laine Yonker/Joe Green |
Managing Director and CEO | XEC Partners | Edison Investor Relations |
+61 (0)3 9827 8999 | ||
1300 932 037 | +1 646-653-7035 |
About Limb Girdle Muscular Dystrophy R2 caused by the genetic loss of dysferlin.
Limb-girdle muscular dystrophy (LGMD) is a group of rare muscular dystrophies primarily characterized by hip and shoulder (i.e. limb-girdle) proximal muscle weakness estimated to occur as a class in up to 6.9 in every 100,000 people5. Over 30 subtypes have been identified with different underlying genetic causes. Dysferlin loss occurs in the recessive genetic muscle disorder LGMDR2, formerly known as LGMD2B, and the related muscle disease Miyoshi myopathy dystrophy 1 (MMD1)6 . Collectively these disorders caused by the loss of dysferlin are called Dysferlinopathy 1. Dysferlinopathy (LGMDR2) prevalence is approximately ~ 1 per 125,000 people3 or 2500 people in the US, or 18% of all LGMDs4 with an estimated upper range for LGMDR2 of ~ 4000 people in the US.
In LGMDR2, the initial weakness is seen in the proximal muscles and in the case of MMD1 there is initially distal muscle weakness. However, natural history data on dysferlinopathy have shown that LGMDR2 and MMD1 are the same disease6. Dysferlin is a transmembrane protein that has been shown to be involved in multiple cellular processes such as calcium regulation, membrane repair, and membrane trafficking. The absence of dysferlin in LGMDR2 is characterized by activated F4/80 macrophage and T cell inflammation and increase in fat replacing the muscle, where adipocytes replace dysferlin deficient muscle, as detected by MRI. This progressively reduces muscle strength, ability to walk, reduces upper limb function and quality of life.5 There are currently no treatments for dysferlinopathy. The steroid drug deflazacort treatment failed in a 6 month patient study and made the disease worse. Deflazacort (1mg) taken for a month, every second day for 5 months reduced muscle strength, which reversed after cessation of steroid treatment7. There is an unmet need for effective therapies so that people with LGMDR2 can maintain ambulation, muscle strength and quality of life.
References
https://www.jain-foundation.org/research/dysferlin-background/therapeutic-opportunities/
Liu W et al Genetics in Medicine 21 2512-2520 (2019) Estimates 0.75 per 100000 https://www.nature.com/articles/s41436-019-0544-8
Moore et al J Neuropathol Exp. Neurol 2006 65(10): 995-1003 https://academic.oup.com/jnen/article/65/10/995/2646712
https://rarediseases.org/rare-diseases/limb-girdle-muscular-dystrophies/
The study results in the psoas muscles above suggest target CD49d RNA knockdown when CD8+ and F4/80+ cells remain in the female psoas muscle. In the quadricep muscles the target CD49d knockdown is less visible as the CD8+ and F4/80+ cells that express CD49d are substantially reduced (by 86% and 70% respectively) so there are less cells remaining to observe a statistically significant reduction versus untreated control mice.
Attachments

